La Administración de Medicamentos y Alimentos de los Estados Unidos (FDA en Inglés) otorgó hoy autorización de uso de emergencia a la primera prueba de COVID-19 para autodiagnóstico en el hogar.
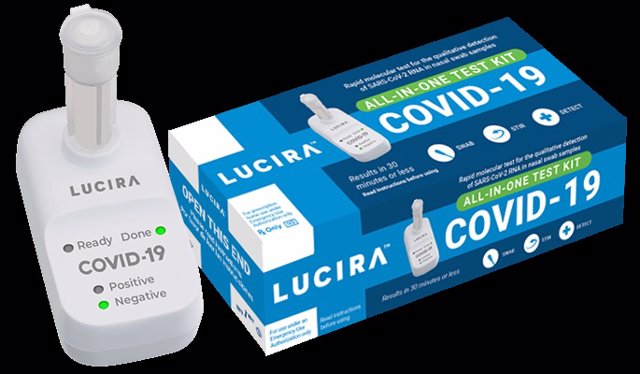
Se trata del kit Lucira COVID-19 All-In-One, una prueba molecular en tiempo real que detecta la presencia de material genético del SARS-CoV-2 en la muestra.
La prueba está dirigida a mayores de 14 años y consiste en introducir el hisopo en una de las fosas nasales, tal y como se hace con las pruebas PCR-RT; luego se debe revolver la muestra con un vial y esperar 30 minutos para obtener el resultado, el cual es anunciado por una pantalla en el dispositivo que forma parte del kit.
Stephen M. Hahn, comisionado de la FDA, afirmó que la institución sigue demostrando una velocidad sin precedentes para responder a la pandemia.
Si bien las pruebas de diagnóstico de COVID-19 han sido autorizadas para la recolección en el hogar, esta es la primera que se puede autoadministrar por completo y proporcionar resultados en el hogar. Esta nueva opción de prueba es un avance diagnóstico importante para abordar la pandemia y reducir la transmisión de la enfermedad.
La FDA indicó que quienes usen esta prueba y salgan positivos, deberán aislarse y buscar atención médica; mientras que quienes den negativo pero tengan síntomas similares a la COVID-19, deberán consultar con su médico ya que los resultados negativos no excluyen que una persona contraiga la infección.
La autorización de uso de emergencia (EUA, por sus siglas en Inglés) permite a la FDA recurrir a productos médicos sin aprobar, o a usos no autorizados de productos médicos que sí están aprobados, para diagnosticar, tratar o prevenir enfermedades o padecimientos graves o potencialmente mortales ocasionados por ciertos agentes peligrosos, siempre y cuando no haya alternativas adecuadas, aprobadas ni disponibles.
Estas autorizaciones fueron promulgadas al amparo de una reforma legal hecha en el año 2013 para la preparación y respuesta a las pandemias. Cuando se declare que la emergencia ha terminado, la autorización de uso de emergencia del producto dejará de ser válida y deberá someterse a los procedimientos usuales de revisión por parte de la FDA.