El medicamento de AstraZeneca logra una reducción del tamaño tumoral de al menos un 20% en el 97% de los pacientes pediátricos tratados durante su primer año.
La neurofibromatosis tipo 1 (NF1) es una enfermedad genética poco frecuente y progresiva que afecta aproximadamente a 1 de cada 3.000 personas en el mundo. Se caracteriza por el desarrollo de tumores no malignos en el cerebro, la médula espinal y los nervios, y suele diagnosticarse en la infancia.
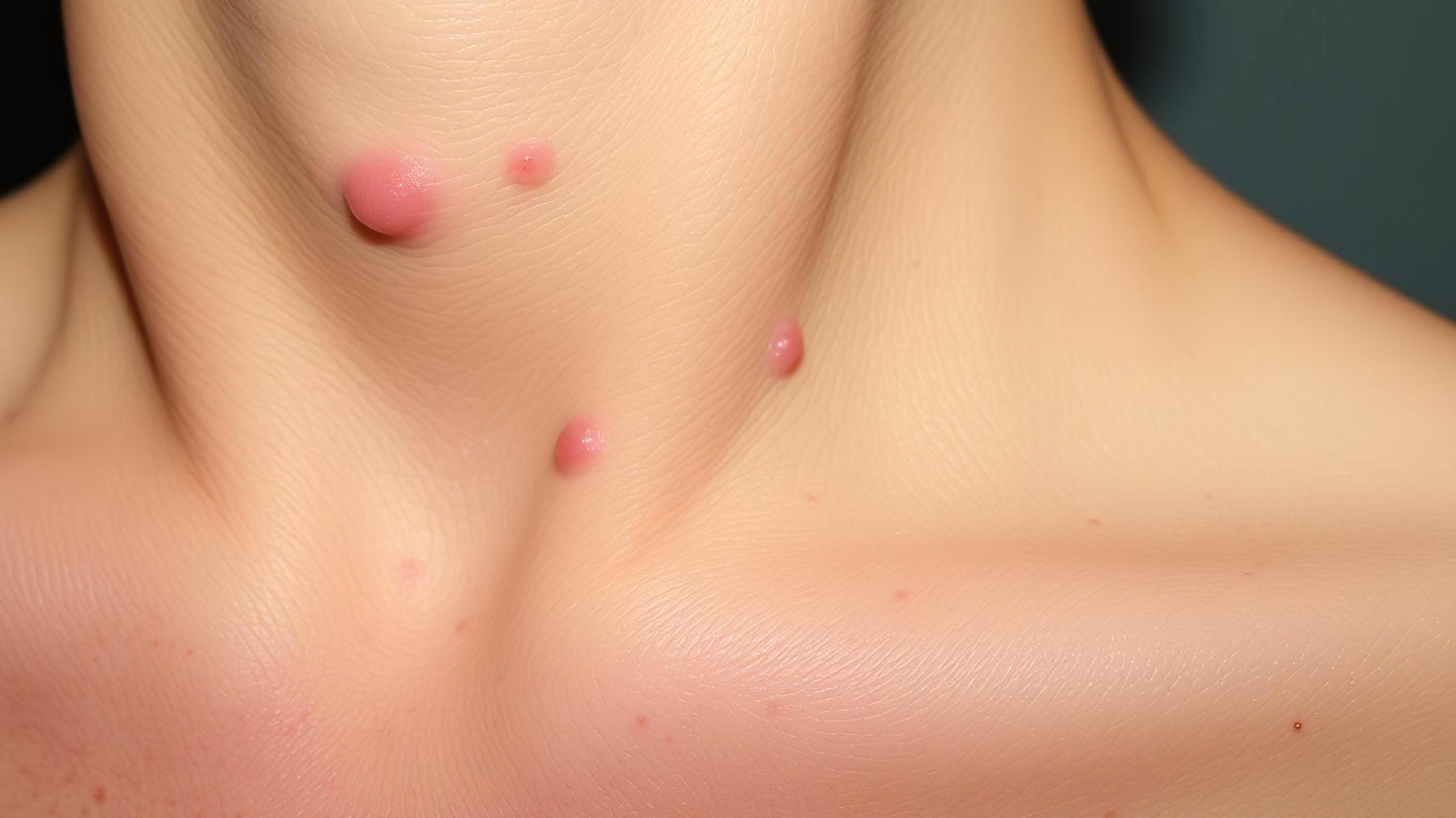
En los niños, uno de los principales riesgos asociados a esta patología es la aparición de neurofibromas plexiformes (NP), tumores que crecen en las vainas nerviosas —tejido que cubre y protege los nervios— y que pueden afectar la movilidad, generar dolor o incluso derivar en complicaciones graves. Alrededor del 10% de estos casos pueden evolucionar a tumores malignos.
Un avance terapéutico para una enfermedad rara
Hasta hace poco, el tratamiento de los neurofibromas plexiformes se enfocaba en aliviar síntomas mediante fisioterapia o control del dolor. Sin embargo, con la aprobación de Koselugo (selumetinib), desarrollada por AstraZeneca, los pacientes pediátricos disponen por primera vez de una opción oral capaz de reducir el tamaño de los tumores inoperables.
Koselugo es el primer medicamento aprobado por la FDA para tratar a niños desde los dos años con NF1 y neurofibromas plexiformes no extirpables.
De acuerdo con los datos clínicos, el 97% de los pacientes tratados con Koselugo experimentaron una reducción de al menos un 20% en el tamaño del tumor entre los 3,3 meses y un año de tratamiento. Además, el 79% mantuvo esta respuesta durante al menos dos años y el 64% por tres años consecutivos.
El Dr. Andrés Rojas, director médico de AstraZeneca Centroamérica y Caribe, comentó:
La investigación en este tipo de enfermedades presenta desafíos únicos que requieren innovación continua. Nos impulsa la vida de los pacientes, y por eso desarrollamos herramientas para comprender mejor estas patologías poco usuales y sus necesidades”.
Reconocer los síntomas y actuar a tiempo
La NF1 puede tener origen hereditario y se debe a una mutación en el gen NF1, responsable de producir la proteína neurofibromina. Aproximadamente el 50% de los casos se presentan sin antecedentes familiares conocidos.
El diagnóstico temprano es fundamental para evitar complicaciones. Entre los signos más comunes de la enfermedad se encuentran:
- Manchas de color marrón claro en la piel
- Bultos suaves sobre o bajo la piel
- Pecas en lugares inusuales
- Gliomas ópticos (tumores alrededor del nervio óptico)
- Escoliosis, problemas óseos o estatura baja
- Retraso o pubertad precoz
- Problemas de aprendizaje o de visión
El diagnóstico puede requerir evaluaciones médicas, estudios de imagen y pruebas genéticas. Actuar con rapidez es esencial, especialmente durante los primeros 10 años de vida, cuando el crecimiento de los tumores puede ser impredecible.
“Es muy importante estar atentos a la presencia de uno o varios síntomas y consultar oportunamente a los especialistas”.
Un compromiso con la ciencia y la vida
La NF1 afecta múltiples aspectos de la vida de los pacientes y sus cuidadores. Por ello, el diagnóstico temprano y un tratamiento integral son clave para mejorar el pronóstico y la calidad de vida.